
Elucidating the Prion-like Propagation and Neurodegenerative Mechanisms of α-Synuclein and Tau Seeds
Research Directions
1. Environmental factors with neurodegenerative disorders
Environmental Factors in Protein Aggregation and Prion-like Strain Evolution. Environmental exposures—including toxins, particulate matter (Science 2025; Biomaterials 2013), bacteria, and viruses (JMV 2024)—have been shown to trigger the aggregation of prion-like proteins. Notably, proteins such as amyloid-β, tau, and α-synuclein, which exhibit prion-like properties, also demonstrate intrinsic antimicrobial activity. Our research focuses on elucidating how exogenous environmental components influence the conformational transformation and subsequent aggregation of these proteins. By comparing the conformational signatures of aggregates derived from patient tissues and those induced by environmental exposures, we aim to better understand the etiology of conformational diseases. This approach enables the identification of shared structural features and environmental inducers that may initiate or accelerate aggregation. Furthermore, uncovering molecular pathways and modifiers of aggregation provides opportunities for the development of early-stage preventive strategies and disease-modifying therapies in neurodegeneration.
Current Projects:
-
Lewy body dementia promotion by air pollutants (Science 2025)
-
SARs-CoV-2 infection in humanized ACE2 mouse model in Alzheimer's disease (Journal of Medical Virology, 2024)



2. Elucidating Prion-like Protein Cell-to-Cell Spreading Mechanisms
-
Our lab is at the forefront of uncovering the pathways facilitating the intercellular transmission of pathological α-synuclein (α-syn) and tau, the prion-like proteins implicated in Parkinson's and Alzheimer's diseases. We have identified several transmembrane proteins, including lymphocyte activation gene-3 (LAG3), amyloid precursor-like protein 1 (APLP1), and neurexins, that specifically bind and internalize these disease-associated protein species.
-
A key finding is that internalized pathological α-syn triggers PARP-1 activation and PAR polymer accumulation, converting it into a highly toxic strain that accelerates neurotoxicity. Remarkably, depleting or inhibiting PARP-1 substantially impedes α-syn transmission and neurodegeneration.
Current Projects:
-
Dissecting the LAG3-APLP1 uptake pathways for therapeutic targeting
-
Interrogating the PARP1/PAR pathway in spreading and identifying inhibitors

-
We identified LAG3, APLP1, and Neurexins as pathogenic α-syn receptors.
-
All these receptors exhibit preferential binding with α-syn preformed fibrils (PFF) compared to α-syn monomer.
-
Depletion of LAG3 or inhibition of LAG3 by anti-LAG3 can inhibit the neuronal uptake of α-syn PFF, subsequent pathology propagation, and neurotoxicity, and alleviate behavioral deficits.
Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson's disease: Lag3 binds with α-syn PFF but not monomer (PNAS 2021)
-
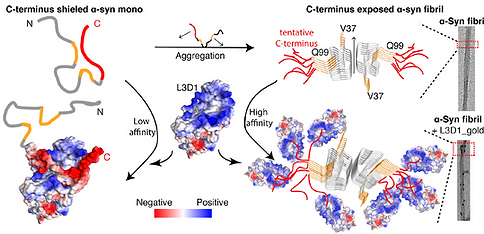
Formation of α-syn fibrils disrupts intramolecular interactions with the C-terminus, exposing and condensing the C-terminus on the fibril surface, increasing binding affinity to the receptors.
-
α-Syn serine 129 phosphorylated (pS129), a pathological α-syn modification, further enhances fibril binding to receptors.
-
pS129 fibrils showed higher cellular internalization, seeding ability, and induced α-syn pathology in transgenic mice.

Aplp1 interacts with Lag3 to facilitate the transmission of pathologic α-syn (Nat Comm 2024)
-
APLP1 is an independent receptor of pathological α-syn, but can also interact with LAG3, facilitating the binding, internalization, transmission, and toxicity of .
-
Deleting both APLP1 and LAG3 can reduce up to 95% α-syn pathology, and eliminate the loss of dopaminergic neurons and related behavioral deficits induced by α-syn PFFs in experimental models.
-
Anti-LAG3 antibodies prevent the internalization of α-syn PFFs by disrupting the APLP1-LAG3 interaction, and block the neurodegeneration induced by α-syn PFFs in vivo.

LAG3 Facilitates Pathological Tau Neuron-to-Neuron Transmission (Adv Sci 2024, back cover)
-
Lag3 is a cell surface receptor that binds specifically to the pathological tau PFF, but not to tau monomer.
-
Deletion or inhibition of Lag3 in neurons significantly reduces the internalization of tau PFF, subsequent tau propagation, and neuron-to-neuron transmission of tau pathology.
-
In mouse models, deletion of neuronal Lag3 attenuates the propagation of tau pathology and behavioral deficits induced by injection of tau PFF in the brain.

Poly (ADP-ribose) drives pathologic α-syn neurodegeneration in Parkinson’s disease (Science 2018)
-
This study reveals that pathologic α-syn activates poly(ADP) polymerase-1 (PARP-1), triggering PAR generation.
-
PAR generation accelerates the formation of pathologic α-syn, resulting in cell death via parthanatos (a type of cell death).
-
Inhibition of PARP-1 either pharmacologically or genetically prevents the toxicity induced by pathologic α-syn.
-
A feed-forward loop is observed where PAR further converts pathologic α-syn into a more toxic strain.
-
Increased levels of PAR are found in the cerebrospinal fluid and brains of PD patients, suggesting PARP activation's role in PD pathogenesis.

Nanotechnology-Based Interventions Against Pathogenic Seed Transmission


3. Developing Therapeutics to Block Prion-like Protein Propagation
-
Building on our mechanistic insights, we are actively pursuing therapeutic strategies to halt the propagation of pathological α-synuclein and tau conformers:
-
Generating and evaluating nanobodies that disrupt prion-like protein aggregation and intercellular transmission (Nat Comm 2022)
-
Leveraging nanozymes to scavenge reactive oxygen species implicated in prion pathology (Nano Today 2021)
-
Through our multidisciplinary approach, combining molecular biology, bioengineering, and nanomedicine, we aim to elucidate key mechanisms and deliver effective therapeutics for prion proteinopathies.